Chapter 7 Identifying confounding factors - Caron set
normPlotDirBit <- "Plots/Norm" # "ConfoundPlots"
#setNameUpp <- "Caron"
#setNameLow <- "caron"
setName <- tolower("Caron")
setSuf <- "_5hCellPerSpl"
typeNorm <- "scran"
projDir <- params$projDir
dirRel <- params$dirRel
outDirBit <- params$outDirBitcaron
7.1 Load object
# Read object in:
tmpFn <- sprintf("%s/%s/Robjects/%s_sce_nz_postDeconv%s.Rds",
projDir, outDirBit, setName, setSuf)
tmpFn## [1] "/ssd/personal/baller01/20200511_FernandesM_ME_crukBiSs2020/AnaWiSce/AnaCourse1/Robjects/caron_sce_nz_postDeconv_5hCellPerSpl.Rds"## class: SingleCellExperiment
## dim: 16629 5500
## metadata(0):
## assays(2): counts logcounts
## rownames(16629): ENSG00000237491 ENSG00000225880 ... ENSG00000275063
## ENSG00000271254
## rowData names(11): ensembl_gene_id external_gene_name ... detected
## gene_sparsity
## colnames: NULL
## colData names(16): Barcode Run ... cell_sparsity sizeFactor
## reducedDimNames(0):
## altExpNames(0):7.2 PCA
Remember scran PCA:
Normalised counts are stored in ‘logcounts’ assay
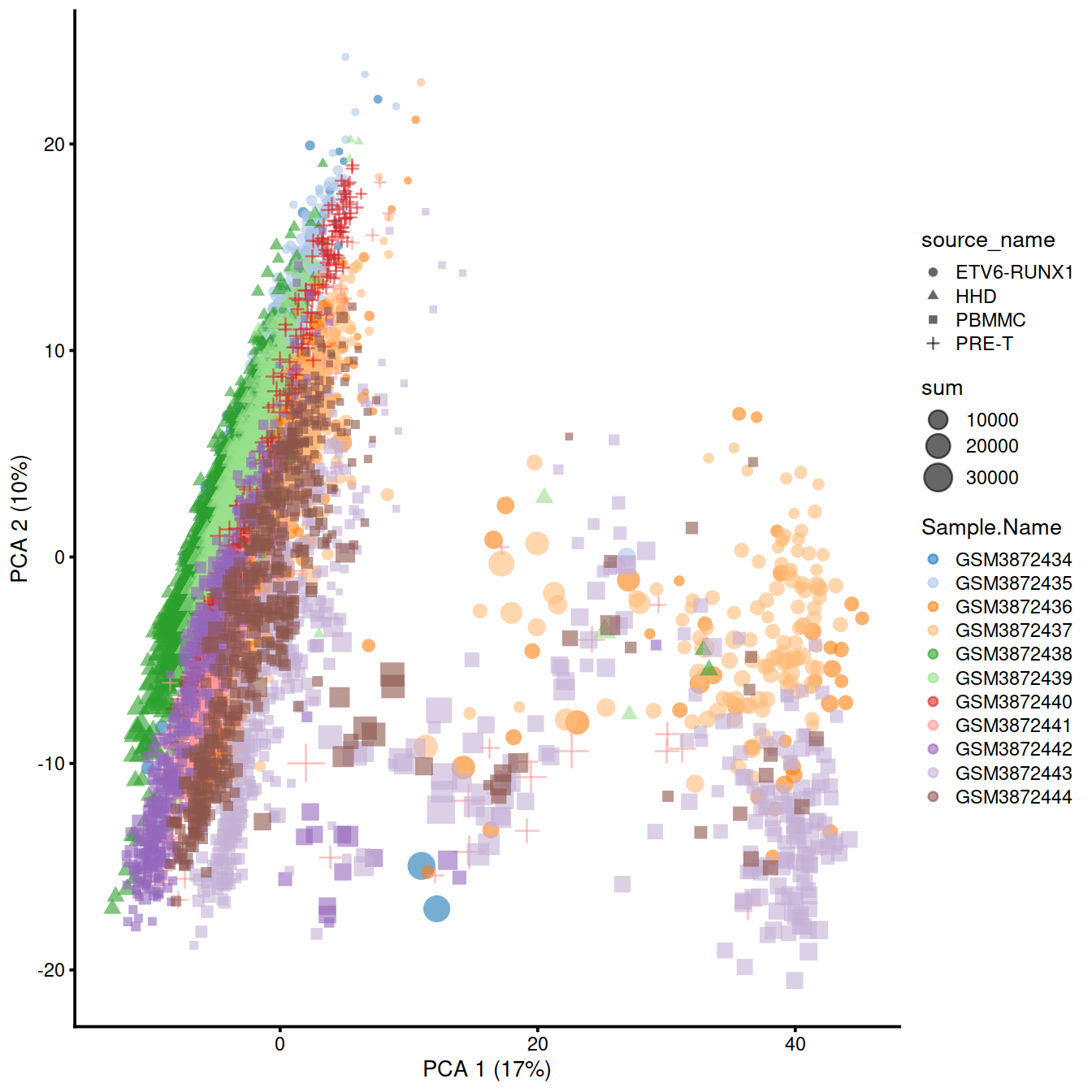
PCA plot for the ‘scran’ counts in the caron set.
tmpFn <- sprintf("%s/%s/%s/%s_sce_nz_postQc%s_%sPca.png",
#projDir, outDirBit, normPlotDirBit, setName, setSuf, typeNorm)
dirRel, normPlotDirBit, setName, setSuf, typeNorm)
tmpFn
knitr::include_graphics(tmpFn, auto_pdf = TRUE)#options(BiocSingularParam.default=IrlbaParam())
options(BiocSingularParam.default=ExactParam())
qclust <- quickCluster(sce, min.size = 30, use.ranks = FALSE)
sce <- computeSumFactors(sce, sizes = 15, clusters = qclust)
sce <- logNormCounts(sce)Perform PCA:
reducedDim(sce, "PCA") <- reducedDim(
runPCA(sce, exprs_values = "logcounts", ncomponents = 10), "PCA")
plotPCA(
sce,
colour_by = "Sample.Name",
size_by = "sum",
shape_by = "source_name"
)
# on norm count https://biocellgen-public.svi.edu.au/mig_2019_scrnaseq-workshop/public/normalization-confounders-and-batch-correction.html#identifying-confounding-factors
# on logcounts_raw https://scrnaseq-course.cog.sanger.ac.uk/website/cleaning-the-expression-matrix.html#correlations-with-pcs
# a bit long
# issue with scale, trying with explanPc/100
# see next chunk too
explanPc <- getExplanatoryPCs(
sce,
exprs_values = "logcounts_raw",
variables = c(
"sum",
"detected",
"source_name",
"Sample.Name",
"subsets_Mito_percent"
)
)
plotExplanatoryPCs(explanPc/100) # on logcounts_raw
# https://biocellgen-public.svi.edu.au/mig_2019_scrnaseq-workshop/public/normalization-confounders-and-batch-correction.html#identifying-confounding-factors
plotExplanatoryVariables(
sce,
exprs_values = "logcounts_raw",
#exprs_values = "counts",
#exprs_values = "logcounts",
variables = c(
"sum",
"detected",
"source_name",
"Sample.Name",
"subsets_Mito_percent"
)
)7.3 Normalised counts
Correlation with PCs: logcounts (normalised):
# on norm count https://biocellgen-public.svi.edu.au/mig_2019_scrnaseq-workshop/public/normalization-confounders-and-batch-correction.html#identifying-confounding-factors
# on logcounts_raw https://scrnaseq-course.cog.sanger.ac.uk/website/cleaning-the-expression-matrix.html#correlations-with-pcs
# a bit long
colData(sce)$source_name <- factor(colData(sce)$source_name)
explanPc <- getExplanatoryPCs(sce,
#exprs_values = "logcounts", # default
variables = c(
"sum",
"detected",
"source_name",
"Sample.Name",
"subsets_Mito_percent"
)
)
plotExplanatoryPCs(explanPc/100) 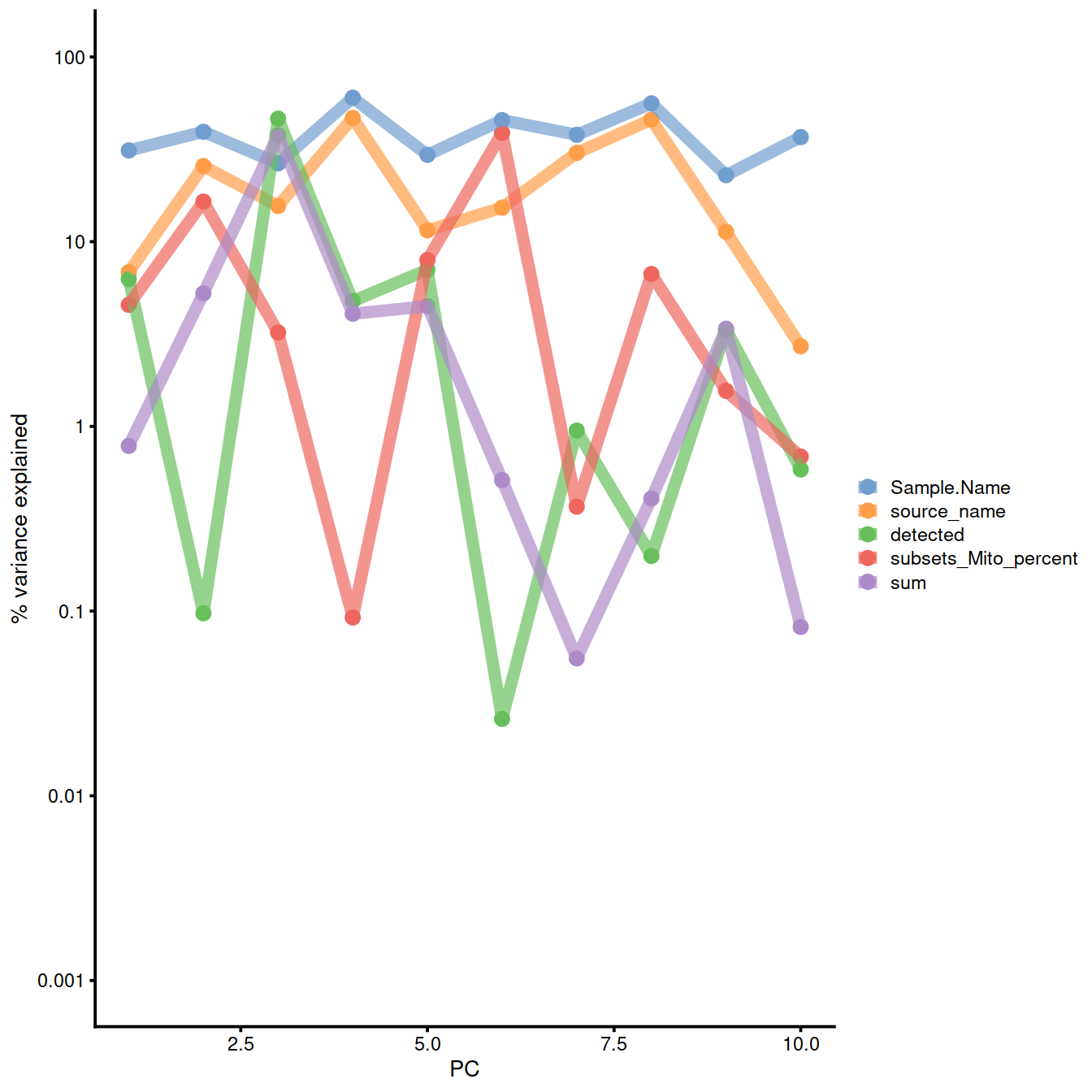
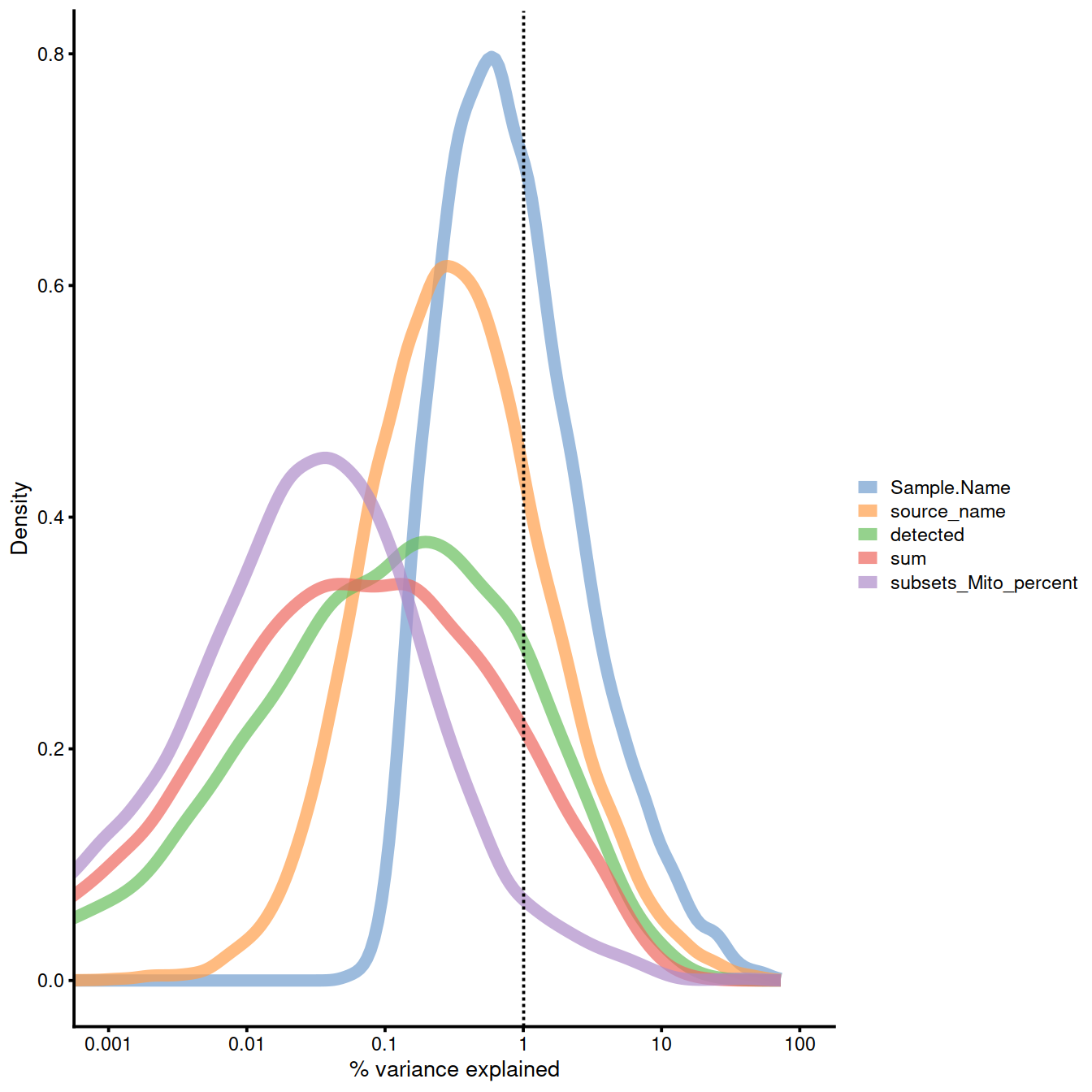
Explanatory variables: logcounts (normalised):
# on logcounts_raw
# https://biocellgen-public.svi.edu.au/mig_2019_scrnaseq-workshop/public/normalization-confounders-and-batch-correction.html#identifying-confounding-factors
plotExplanatoryVariables(
sce,
# exprs_values = "logcounts", # default
variables = c(
"sum",
"detected",
"source_name",
"Sample.Name",
"subsets_Mito_percent"
)
)
7.4 Session information
## R version 4.0.3 (2020-10-10)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: CentOS Linux 8
##
## Matrix products: default
## BLAS: /opt/R/R-4.0.3/lib64/R/lib/libRblas.so
## LAPACK: /opt/R/R-4.0.3/lib64/R/lib/libRlapack.so
##
## locale:
## [1] LC_CTYPE=en_GB.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_GB.UTF-8 LC_COLLATE=en_GB.UTF-8
## [5] LC_MONETARY=en_GB.UTF-8 LC_MESSAGES=en_GB.UTF-8
## [7] LC_PAPER=en_GB.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_GB.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] parallel stats4 stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] Cairo_1.5-12.2 BiocSingular_1.6.0
## [3] dplyr_1.0.6 scran_1.18.7
## [5] scater_1.18.6 ggplot2_3.3.3
## [7] SingleCellExperiment_1.12.0 SummarizedExperiment_1.20.0
## [9] Biobase_2.50.0 GenomicRanges_1.42.0
## [11] GenomeInfoDb_1.26.7 IRanges_2.24.1
## [13] S4Vectors_0.28.1 BiocGenerics_0.36.1
## [15] MatrixGenerics_1.2.1 matrixStats_0.58.0
## [17] knitr_1.33
##
## loaded via a namespace (and not attached):
## [1] viridis_0.6.1 edgeR_3.32.1
## [3] sass_0.4.0 jsonlite_1.7.2
## [5] viridisLite_0.4.0 DelayedMatrixStats_1.12.3
## [7] scuttle_1.0.4 bslib_0.2.5
## [9] assertthat_0.2.1 statmod_1.4.36
## [11] highr_0.9 dqrng_0.3.0
## [13] GenomeInfoDbData_1.2.4 vipor_0.4.5
## [15] yaml_2.2.1 pillar_1.6.1
## [17] lattice_0.20-44 limma_3.46.0
## [19] glue_1.4.2 beachmat_2.6.4
## [21] digest_0.6.27 XVector_0.30.0
## [23] colorspace_2.0-1 cowplot_1.1.1
## [25] htmltools_0.5.1.1 Matrix_1.3-3
## [27] pkgconfig_2.0.3 bookdown_0.22
## [29] zlibbioc_1.36.0 purrr_0.3.4
## [31] scales_1.1.1 BiocParallel_1.24.1
## [33] tibble_3.1.2 farver_2.1.0
## [35] generics_0.1.0 ellipsis_0.3.2
## [37] withr_2.4.2 magrittr_2.0.1
## [39] crayon_1.4.1 evaluate_0.14
## [41] fansi_0.4.2 bluster_1.0.0
## [43] beeswarm_0.3.1 tools_4.0.3
## [45] lifecycle_1.0.0 stringr_1.4.0
## [47] locfit_1.5-9.4 munsell_0.5.0
## [49] DelayedArray_0.16.3 irlba_2.3.3
## [51] compiler_4.0.3 jquerylib_0.1.4
## [53] rsvd_1.0.5 rlang_0.4.11
## [55] grid_4.0.3 RCurl_1.98-1.3
## [57] BiocNeighbors_1.8.2 igraph_1.2.6
## [59] labeling_0.4.2 bitops_1.0-7
## [61] rmarkdown_2.8 codetools_0.2-18
## [63] gtable_0.3.0 DBI_1.1.1
## [65] R6_2.5.0 gridExtra_2.3
## [67] utf8_1.2.1 stringi_1.6.1
## [69] ggbeeswarm_0.6.0 Rcpp_1.0.6
## [71] vctrs_0.3.8 tidyselect_1.1.1
## [73] xfun_0.23 sparseMatrixStats_1.2.1