crukci-cluster-transition
CRUKCI Cluster Transition - Hands-on training
Session 1: Shell
Learning Objectives
- Use most useful shell commands
- Organise data into folders
- Log in onto the cluster
- Understand directories’ structure
- View reference genomes
- Retrieve sequencing data
- Merge datasets
Basics, most useful shell commands
Open a Terminal window:
Syntax
Remember the UNIX/LINUX command line is case sensitive!
-
$indicates start of command -
#indicates end of command and start of comment
The $ and # symbols are not part of the command.
<...> or my_... refers to variables and file names that need to be specified by the user. The arrows <...> need to be excluded, because they are generic UNIX redirection functions!
Orientation
$ whoami # shows as who you are logged in
$ hostname # shows on which machine you are
$ pwd # present working directory
$ ls # list directory contents of pwd
$ ls -F # list directory contents of pwd with an added slash (`/') immediately after each pathname that is a directory
$ ls -l # provides additional info on files and directories
$ ls -la # includes hidden files (.name) as well
$ ls -lat # lists files in chronological order
$ ls -R # lists subdirectories recursively
$ man ls # manual on command ls
$ cd <dir_name> # changes/switches into specified directory
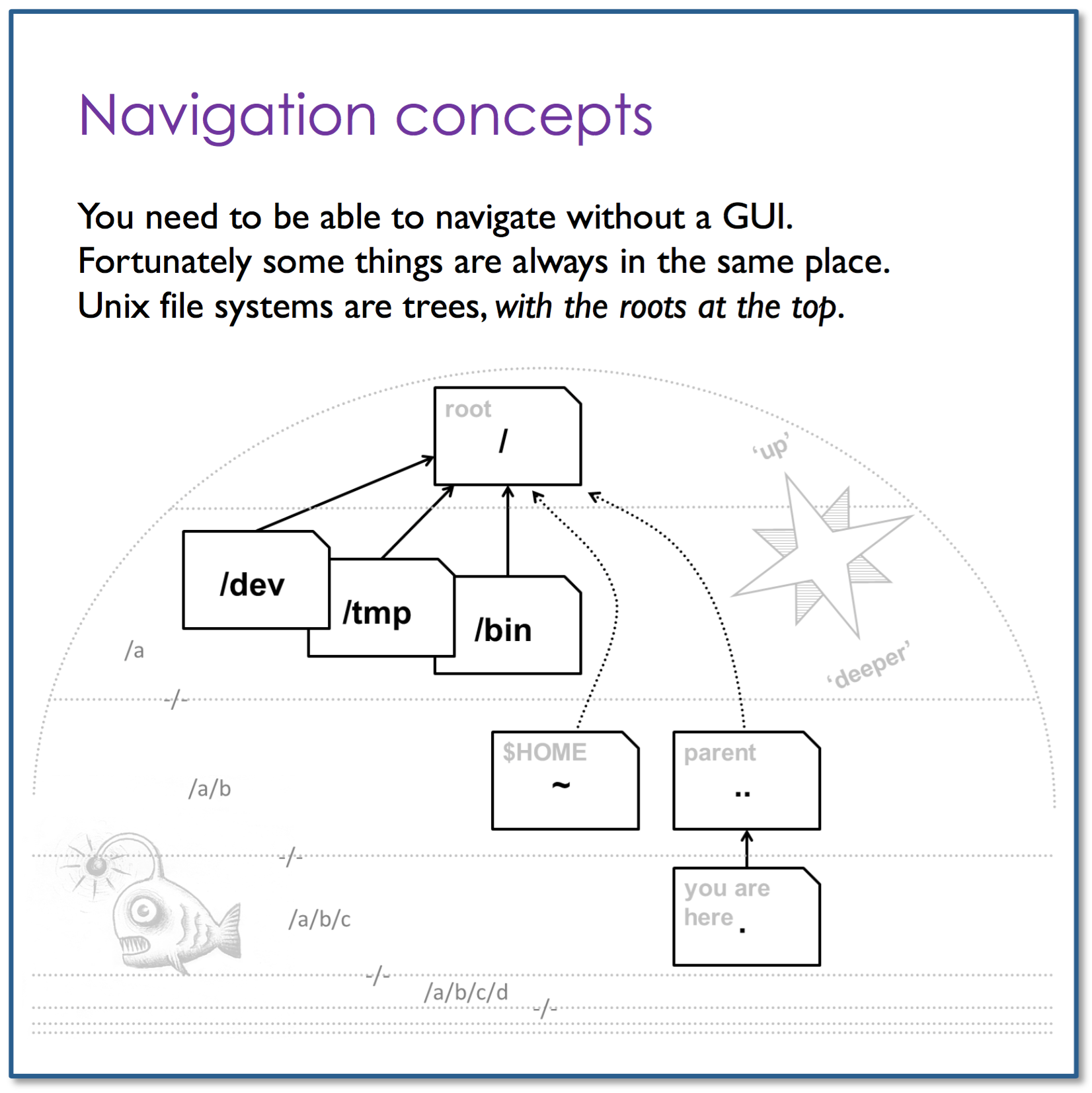
$ cd .. # moves one directory up
$ cd ../../ # moves two directories up (and so on)
$ cd # brings you to highest level of your home directory
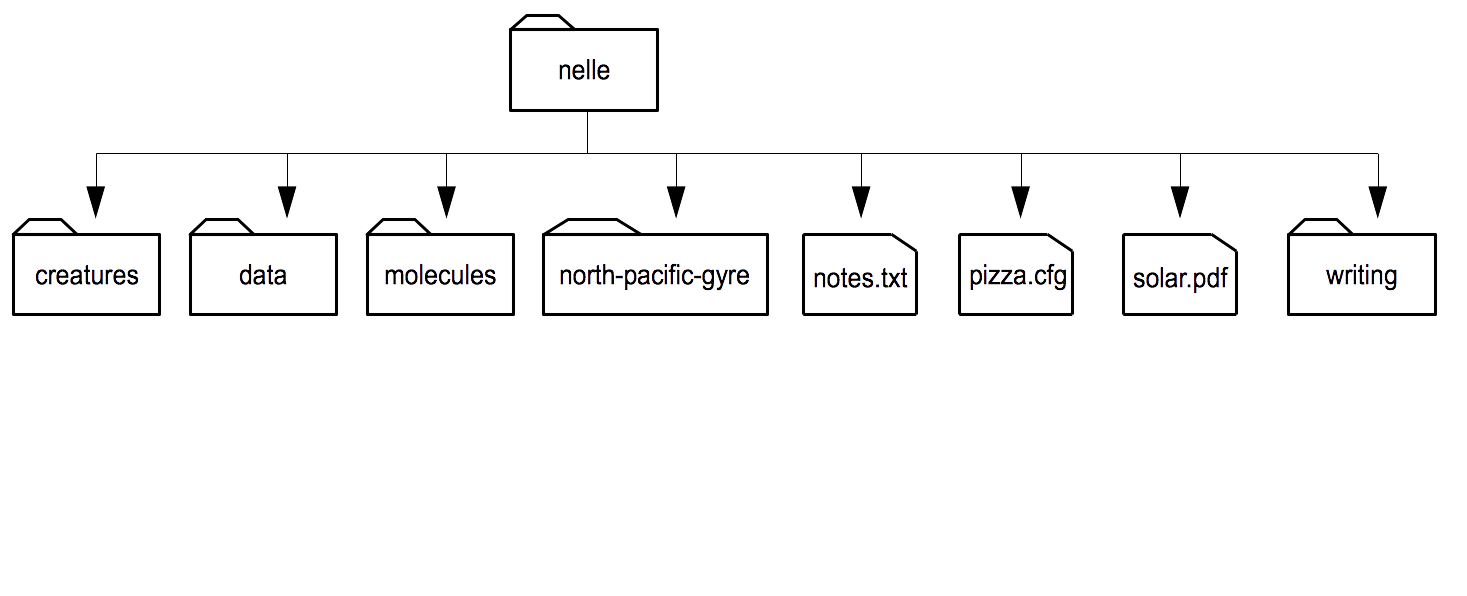
EXERCISE Download Session 1 data , save file onto your computer and unzip it. The downloaded data contains Nelle Nemo’s home directory. Nelle Nemo is a marine biologist, she has just returned from a six-month survey of the North Pacific Gyre, where she has been sampling gelatinous marine life in the Great Pacific Garbage Patch.
Open a Terminal window, navigate to the downloaded file and enter into its contents, go to
nelledirectory. Draw a diagram ofnelle’s directories and files structure. Do not use a graphical interface, only the command line please!
Congratulations!
You did it!
See images/homedir.png for the answer
{kind=link}
Files and directories
Tips on file names:
-
Do not use spaces
- other options include: underscore (
my_data.txt), dashes (my-data.txt), no separation (mydata.txt) or camel case (MyData.txt)
- other options include: underscore (
- Avoid special characters
~ ! @ # $ % ^ & * ( ) ; < > ? , [] {} ' " | - Use short file names
- Use a good format for dates like
YYYYMMDD, then all your files will always stay in chronological order - Add leading zeros for clarity and to make sure files sort in sequential order, for example
001, 002, ... 010, 011, ...
$ mkdir <dir_name> # creates specified directory
$ rmdir <dir_name> # removes empty directory
$ nano <file_name> # create a file using the text editor nano
$ rm <file_name> # removes file name
$ rm -r <dir_name> # removes directory including its content, but asks for confirmation, 'f' argument turns confirmation off
$ mv <name1> <name2> # renames directories or files
$ mv <name> <path> # moves file/directory as specified in path
$ cp <name> <path> # copy file/directory as specified in path (-r to include content in directories)
When we say, “nano is a text editor”, we really do mean “text”: it can only work with plain character data, not tables, images, or any other human-friendly media. We use it in examples because almost anyone can drive it anywhere without training, but please use something more powerful for real work. On Unix systems (such as Linux and Mac OS X), many programmers use Emacs or Vim (both of which are completely unintuitive, even by Unix standards), or a graphical editor such as Atom which is not available from the cluster.
session1-data/nelle
- Create a new directory
thesisand create a filedraft.txtin it. Type this lineIt's not "publish or perish" any more, it's "share and thrive"!if you wish and save.- Navigate back to
session1-data/nelleand deletethesisdirectory.- Let’s create that directory and file one more time.
draft.txtisn’t a particularly informative name, so let’s change it toquotes.txt- Let’s now move
quotes.txtintonelledirectory- Copy
quotes.txtintothesisdirectory asquotations.txtfile
![]() With Great Power Comes Great Responsibility. When the files and directories are deleted, there is no way back. They are totally gone forever.
With Great Power Comes Great Responsibility. When the files and directories are deleted, there is no way back. They are totally gone forever.
Copy and paste
Depends on local environment. Usually one of the following methods works:
- Copy: Ctrl&Shift&c or right/middle mouse click
- Paste: Ctrl&Shift&p or right/middle mouse click
Handy shortcuts
$ ~/ # refers to user's home directory
$ history # shows all commands you have used recently
$ !<number> # starts an old command by providing its ID number
$ up(down) key # scrolls through command history
$ <incomplete path/file_name> TAB # completes path/file_name
$ <incomplete command> SHIFT&TAB # completes command
$ Ctrl a # cursor to beginning of command line
$ Ctrl e # cursor to end of command line
$ Ctrl c # aborts the execution of a command
Filters and pipes
Now that we know a few basic commands, we can finally look at the shell’s most powerful feature: the ease with which it lets us combine existing programs in new ways. We’ll start with a directory called molecules that contains six files describing some simple organic molecules. The .pdb extension indicates that these files are in Protein Data Bank format, a simple text format that specifies the type and position of each atom in the molecule. Which of these molecules have the smallest number of atoms?
$ wc -l <file_name> # counts the number of lines in each input file
$ ls *.txt # list files with name matching zero or more characters and ends with .txt
When the shell sees a wildcard, it expands the wildcard to create a list of matching filenames before running the command that was asked for.
session1-data/nelle/molecules. So, which of these molecules have the smallest number of atoms?
- First, list only the molecules which start with the letter
p. How many are they?- Now, get the numbers of lines for each molecule. Can you do it with only one command?
Which of these molecules have the smallest number of atoms? It’s an easy question to answer when there are only six files, but what if there were 6000? We will redirect the output of the command wc -l into a file instead of printing it to the screen using the greater than symbol >.
$ wc -l <file_name> > length.txt # redirects the command's output to a file length.txt
$ cat <file_name_a> <file_name_b> # prints the contents of files one after another
$ sort -n <file_name> # sorts lines of text file in numerical order instead of alphabetical
$ head -1 <file_name> # gets the first line of the file, use -4 to get the first 4 and so on
Instead of creating enormous programs that try to do many different things, Unix programmers focus on creating lots of simple tools that each do one job well, and that work well with each other. This programming model is called “pipes and filters”.
We’ve already seen filters; a filter is a program like wc or sort that transforms a stream of input into a stream of output. Almost all of the standard Unix tools can work this way: unless told to do otherwise, they read from standard input, do something with what they’ve read, and write to standard output.
To combine these programs, we use the vertical bar between two commands which is called a pipe |. It tells the shell that we want to use the output of the command on the left as the input to the command on the right.
$ wc -l <file_names> | sort -n # standard output of wc is fed directly to the standard input of sort
session1-data/nelle/north-pacific-gyre/2012-07-03. Nelle has 300 samples in all, and ran each sample through an assay machine that will measure the relative abundance of 300 different proteins. The machine’s output for a single sample is a file with one line for each protein.
- How many assay result files does she have?
- Could you check that each file has 300 measurements as expected?
Navigation recap
Connecting to Cluster
Moving data, or yourself
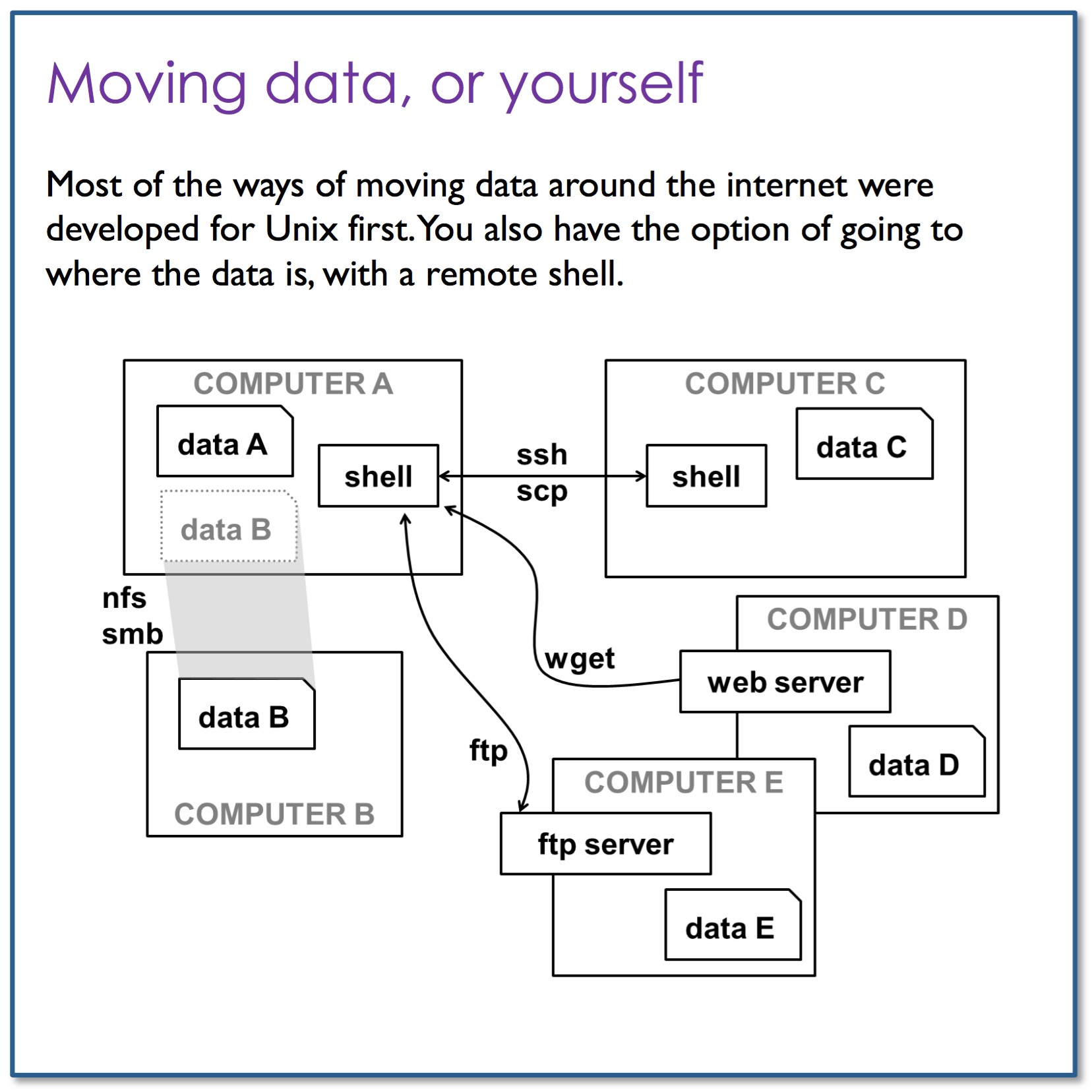
Accessing Remote Server
ssh (Secure Shell) is a network protocol that allows a secure access over an encrypted connection. Through an SSH connection you can easily manage your files and folders, modify their permissions, edit files directly on the server, configure and install your scripts, etc. ssh is used to securely login to a Linux / UNIX host running the sshd daemon on a reachable network.
First, you need a cluster account, request one via Helpdesk - IT ithelpdesk@cruk.cam.ac.uk.
Access the cluster at clust1-headnode.cri.camres.org using the ssh command and your username, press enter and type your password.
$ ssh my_username@clust1-headnode.cri.camres.org
At this point, you are physically on the cluster head node inside your home directory. The directory structure is now completely different to the one you had before and you can no-longer access the files within your file system. However, you can still use the same commands you have seen already. If you now type these commands the outputs will now be specific to you and to the server your are running them on. To return to your computer, use the command exit.
$ exit
- What is the full path of your home directory?
- Do you have any files? How many?
- Return to your own computer, make sure your hostname is your own machine.
Cluster directories’ structure
- Home directory:
/home/$username - Shared installed software:
/home/bioinformatics/software - Scratch/working areas:
/scratchaand/scratchb - Reference data:
/scratchb/bioinformatics/reference_data
The two scratch/working areas are separate but equivalent. These areas are (deliberately) not backed, they are a massively parallel distributed file system called Lustre. Pick one to do your work, and to make best use vary which one to use.
Large files may need to be stripped to improve performance or files that many jobs reads. See lfs setstripe --help for help.
Limit number of files in directory, if possible avoid 10,000s files in single directory.
- Go to the bioinformatics shared installed software and list what’s installed.
- Go to one of the scratch spaces to find your lab folder.
- Create a directory under your lab folder in the scratch area using your username for its name e.g.
/scratchb/bioinformatics/pajon01
Reference genomes
- Path to reference genomes:
/scratchb/bioinformatics/reference_data/reference_genomes/ - Path to assembly:
/scratchb/bioinformatics/reference_data/reference_genomes/$organism/$assemblye.g. for Human GRCh38/scratchb/bioinformatics/reference_data/reference_genomes/homo_sapiens/GRCh38
What Bioinformatics Core maintains:
- Genome sequence (fasta)
- Alignment indices: BWA, TopHat, Bowtie (1, 2)
- Annotations:
- GTF format gene model
- RefFlat format gene model
- Signal artifact list (if available)
- Go to the bioinformatics shared reference genomes and list the assemblies installed for Homo Sapiens.
Getting sequencing data
Downloading Files
The wget utility is the best option to download files from the internet. It can pretty much handle all complex download situations including large file downloads, recursive downloads, non-interactive downloads, multiple file downloads etc. It retrieves files from World Wide Web (WWW) using widely used protocols like HTTP, HTTPS and FTP, and is designed in such way so that it works in slow or unstable network connections. wget can automatically re-start a download where it was left off in case of network problem. Also it downloads file recursively and will keep trying until file has be retrieved completely.
$ wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR000/SRR000731/SRR000731.fastq.gz
- Go to your directory under your lab folder in the scratch area e.g.
/scratchb/bioinformatics/pajon01- Download this FASTQ file ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR000/SRR000731/SRR000731.fastq.gz
Copying Files and Directories
ssh protocol can also be used to copy files & directories, using the same connection method as above but the command we use is called scp.
scp -r molecules/ my_username@clust1-headnode.cri.camres.org:/scratchb/xxlab/my_username/
session1-data/nelle.
- Copy the
moleculesdirectory into your directory under your lab folder in the scratch area on the cluster.- Log in to the cluster and navigate to your scratch folder. Check that the molecule files are present.
Using CRUKCI infrastructure
The Bioinformatics Core provides a tool for downloading files for projects, libraries and runs that you can use from the command line or integrate into your Java application. This is available internally from: http://intranet.cri.camres.org/core-facilities/bioinformatics/sequencing/api
Save this file to your working area. You can run the tool from the command line with:
$ wget http://internal-bioinformatics.cruk.cam.ac.uk/software/clarity-tools.jar
$ java -jar clarity-tools.jar --help
To download all sequencing files associated to your SLX-14572, you can use this command:
$ java -jar /path/to/clarity-tools.jar -l SLX-14572
- Download Clarity tool in your home directory
- Navigate to your scratch folder.
- Download your preferred project data.
Merging datasets
To merge or concatenate multiple FASTQ files, you do not need to decompress them. It can be done using the cat command and redirecting its output to a new file.
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR056/ERR056477/ERR056477.fastq.gz
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR056/ERR056478/ERR056478.fastq.gz
cat ERR056477.fastq.gz ERR056478.fastq.gz > PRJEB2772.fastq.gz
- Navigate to your scratch folder.
- Download two FASTQ files and merge them.
Take home message: everyday commands
$ pwd # present working directory
$ ls # list directory contents of pwd
$ cd <dir_name> # changes/switches into specified directory
$ mkdir <dir_name> # creates specified directory
$ nano <file_name> # create a file using the text editor nano
$ cat <file_name> # prints the contents of files one after another
$ rm <file_name> # removes file name
$ rm -r <dir_name> # removes directory including its content, but asks for confirmation, 'f' argument turns confirmation off
$ mv <name1> <name2> # renames directories or files
$ mv <name> <path> # moves file/directory as specified in path
$ cp <name> <path> # copy file/directory as specified in path (-r to include content in directories)
$ wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR056/ERR056477/ERR056477.fastq.gz # download files from the internet
$ scp -r molecules/ clust1-headnode:/scratchb/xxlab/my_username/ # copy files & directories between different computers
$ ssh my_username@clust1-headnode.cri.camres.org # access a remote computer
$ exit # exit ;)