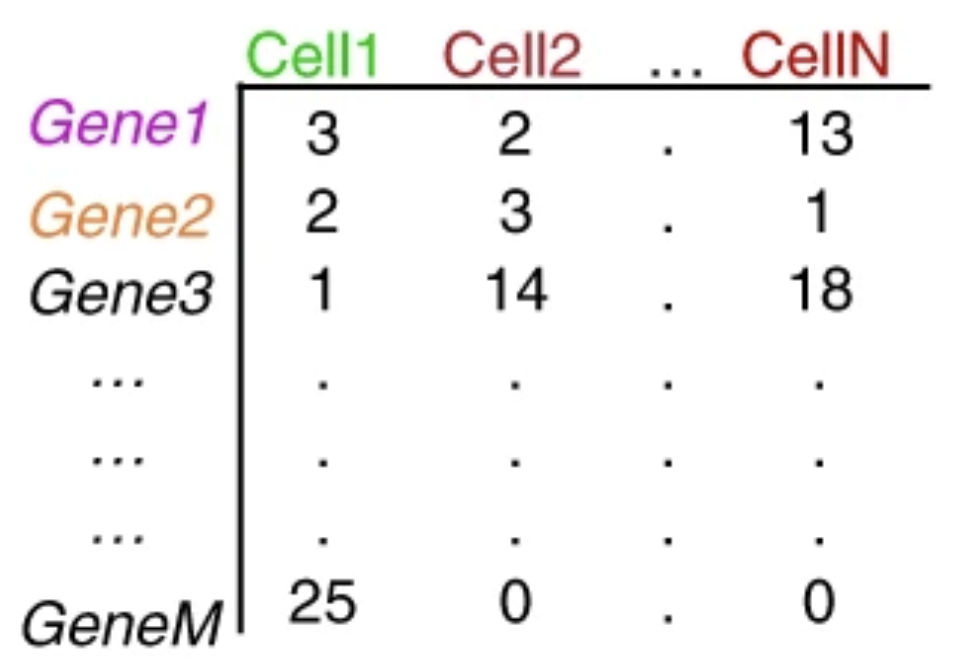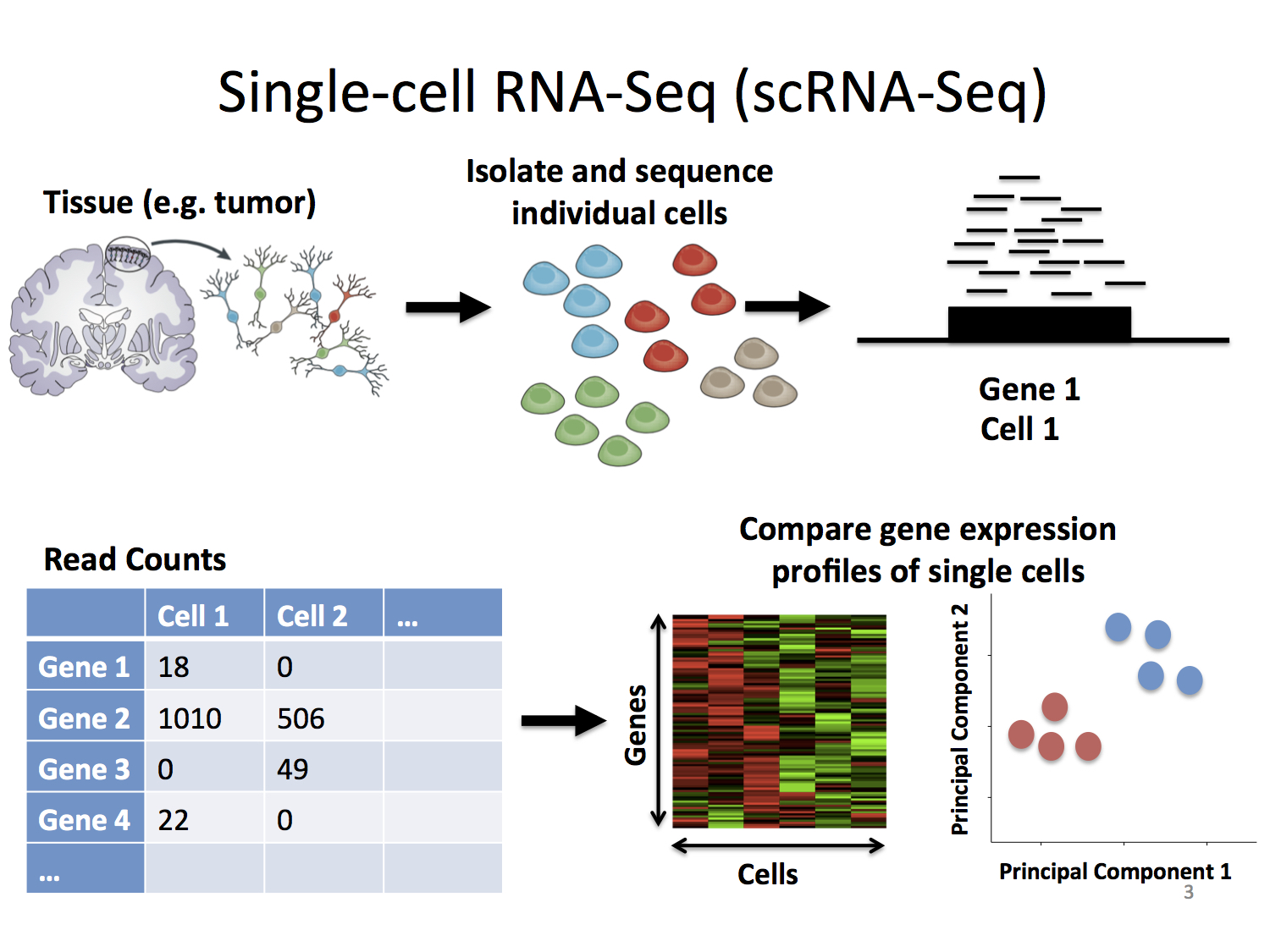
Image by Stephanie Hicks via learn.gencore.bio.nyu.edu
September 2022
Image by Stephanie Hicks via learn.gencore.bio.nyu.edu

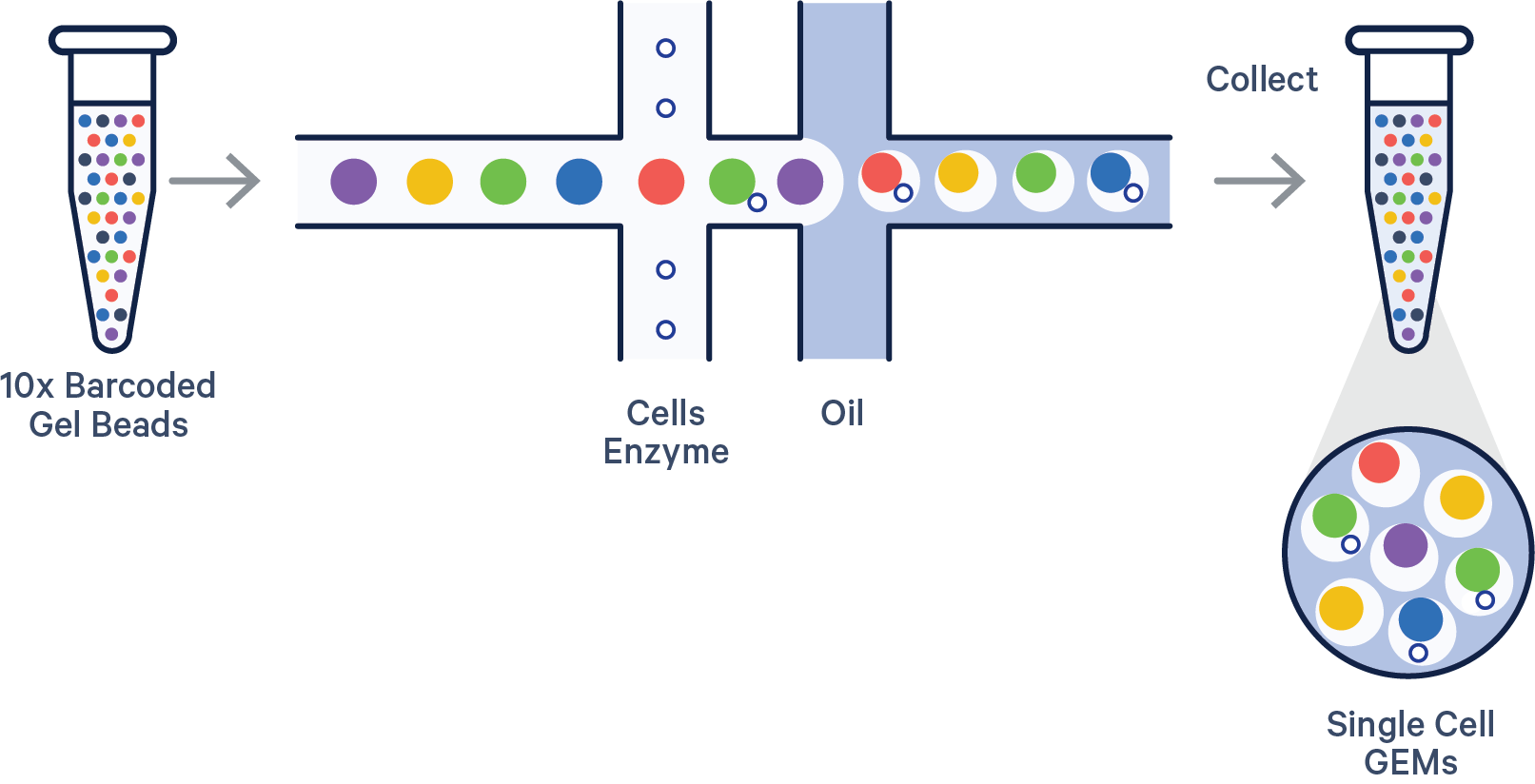
Image by 10x Genomics
The 10x library contains four pieces of information, in the form of DNA sequences, for each “read”.

The sequences for any given fragment will generally be delivered in 3 or 4 files:
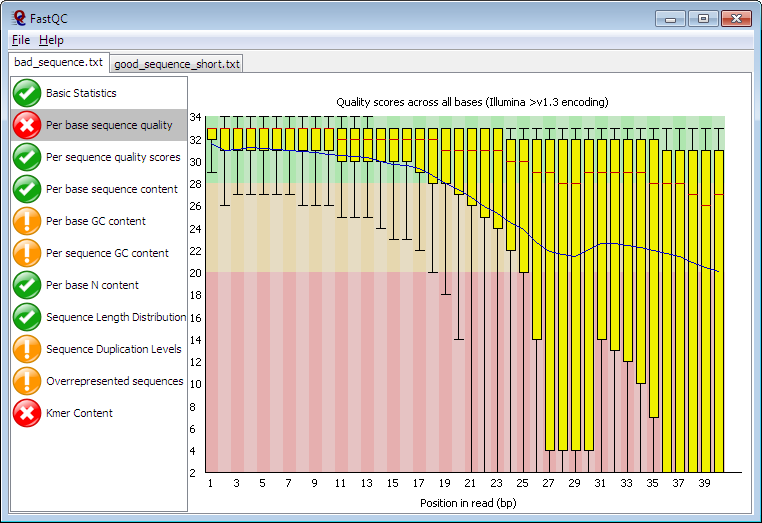


The first steps in the analysis of single cell RNAseq data:
Alternative methods include:

Setup instructions given in the course materials homepage.
Cell Ranger includes a number of different tools for analysing scRNAseq data, including:
cellranger mkref - for making custom referencescellranger count - for aligning reads and generating a count matrixcellranger aggr - for combining multiple samples and normalising the countsCell Ranger requires the fastq file names to follow a convention:
<SampleName>_S<SampleNumber>_L00<Lane>_<Read>_001.fastq.gz
e.g. for a single sample in the Caron data set we have:
SRR9264343_S0_L001_I1_001.fastq.gz
SRR9264343_S0_L001_R1_001.fastq.gz
SRR9264343_S0_L001_R2_001.fastq.gz
As with other aligners Cell Ranger requires the information about the genome and transcriptome of interest to be provided in a specific format.
cellranger mkrefcellranger mkref \
--fasta={GENOME FASTA} \
--genes={ANNOTATION GTF} \
--genome={OUTPUT FOLDER FOR INDEX} \
--nthreads={CPUS}
cellranger countcellranger count \
--id={OUTPUT_SAMPLE_NAME} \
--transcriptome={DIRECTORY_WITH_REFERENCE} \
--fastqs={DIRECTORY_WITH_FASTQ_FILES} \
--sample={NAME_OF_SAMPLE_IN_FASTQ_FILES} \
--localcores={NUMBER_OF_CPUS} \
--localmem={RAM_MEMORY}








Two types of outputs:
.tsv and .mtx.h5Both of these can be read by standard scRNA-seq analysis packages and contain data for a
unique molecular identified (UMI) count matrix: