Introduction to single-cell RNA-seq analysis
Clustering
April 2024
1 Overview
Some of the materials originate in the Hemberg group course material with some of the text copied with a few edits. Also see the OSCA book’s “Basic” and “Advanced” chapters on clustering. In particular, please read the overview with regard to the comments on the “correctness” of any given clustering result.
Once we have normalized the data and removed confounders we can carry out analyses that are relevant to the biological questions at hand. The exact nature of the analysis depends on the data set. One of the most promising applications of scRNA-seq is de novo discovery and annotation of cell-types based on transcription profiles. This requires the identification of groups of cells based on the similarities of the transcriptomes without any prior knowledge of the label a.k.a. unsupervised clustering. To avoid the challenges caused by the noise and high dimensionality of the scRNA-seq data, clustering is performed after feature selection and dimensionality reduction. For data that has not required batch correction this would usually be based on the PCA output. As our data has required batch correction we will use the “corrected” reducedDims data.
We will focus here on graph-based clustering, however, it is also possible to apply hierarchical clustering and k-means clustering on smaller data sets - see the OSCA book for details. Graph-base clustering is a more recent development and better suited for scRNA-seq, especially large data sets.
2 Load the data
library(scater)
library(scran)
library(bluster)
library(cluster)
library(igraph)
library(pheatmap)
library(patchwork)
library(tidyverse)We will use the data set generated in the previous session. This contains 7 samples from the Caron data set. For the purposes of these materials, in the interests of time, each sample has been downsampled to only contain 500 cells.
sce <- readRDS("R_objects/Caron_batch_corrected.500.rds")table(sce$SampleName)##
## ETV6-RUNX1_1 ETV6-RUNX1_2 ETV6-RUNX1_3 ETV6-RUNX1_4 PBMMC_1 PBMMC_2
## 500 500 500 500 500 500
## PBMMC_3
## 5003 Graph-based clustering overview
Graph-based clustering entails building a nearest-neighbour (NN) graph using cells as nodes and their similarity as edges, then identifying ‘communities’ of cells within the network. A graph-based clustering method has three key parameters:
- How many neighbors are considered when constructing the graph
- What scheme is used to weight the edges
- Which community detection algorithm is used to define the clusters
3.1 Connecting nodes (cells) based on nearest neighbours
Two types of NN graph may be used: “K nearest-neighbour” (KNN) and “shared nearest-neighbour” (SNN). In a KNN graph, two nodes (cells), say A and B, are connected by an edge if the distance between them is amongst the k smallest distances from A to other cells. In an SNN graph A and B are connected if the distance is amongst the k samllest distances from A to other cells and also among the k smallest distance from B to other cells.

In the figure above, if k is 5, then A and B would be connected in a KNN graph as B is one of the 5 closest cells to A, however, they would not be connected in an SNN graph as B has 5 other cells that are closer to it than A.
The value of k can be roughly interpreted as the anticipated
size of the smallest subpopulation” (see scran’s
buildSNNGraph() manual).
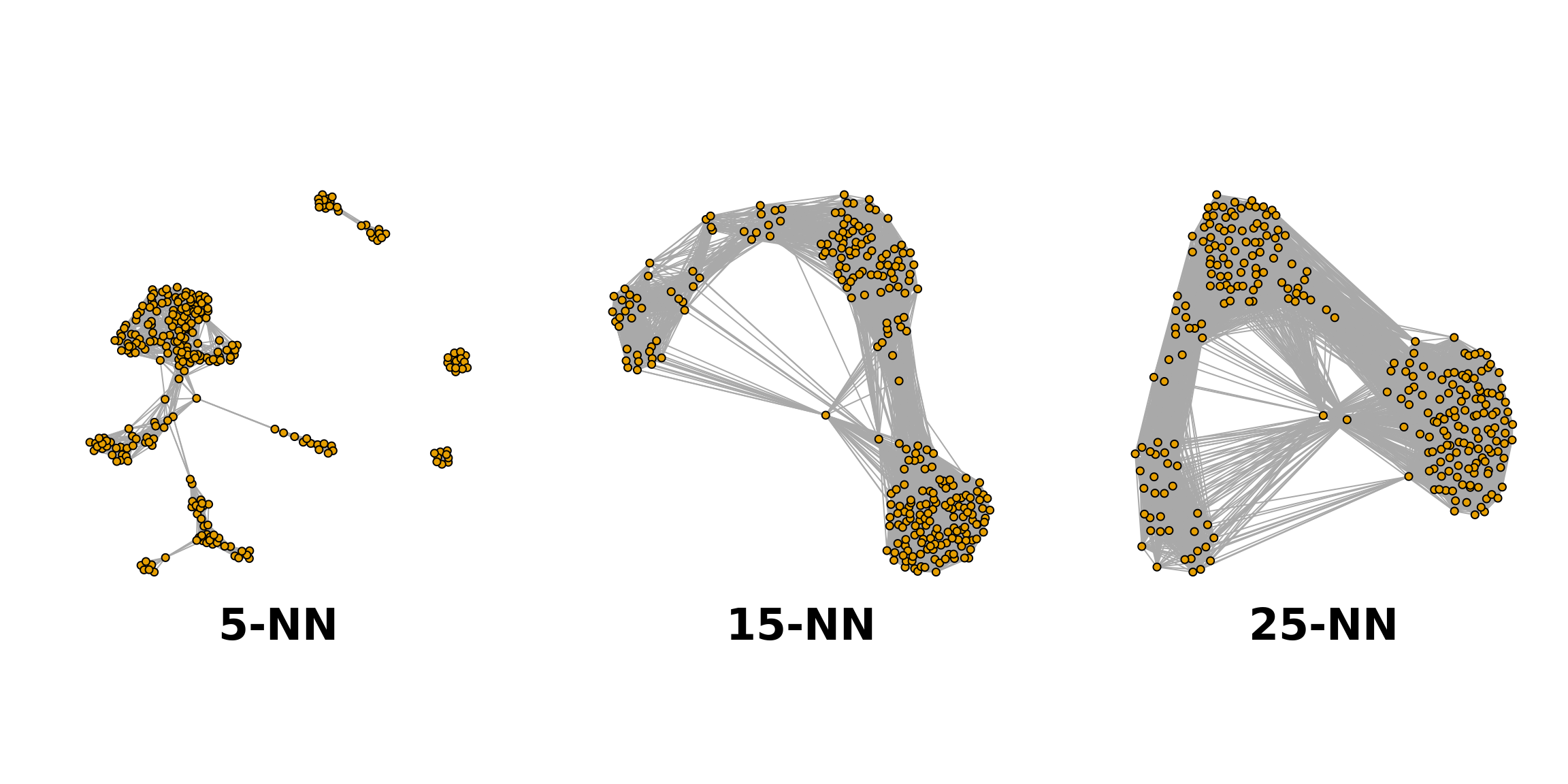
The plot below shows the same data set as a network built using three different numbers of neighbours: 5, 15 and 25 (from here).
3.2 Weighting the edges
The edges between nodes (cells) can be weighted based on the
similarity of the cells; edges connecting cells that are more closely
related will have a higher weight. The three common methods for this
weighting are (see the
bluster package documentation for the makeSNNGraph
function):
- rank - the weight is based on the highest rank of
the shared nearest neighbours
- number - the weight is based the number of nearest
neighbours in common between the two cells
- jaccard - the Jaccard index of the two cells’ sets of nearest neighbours.
3.3 Grouping nodes (cells) into clusters
Clusters are identified using an algorithm that interprets the connections of the graph to find groups of highly interconnected cells. A variety of different algorithms are available to do this, in these materials we will focus on three methods: walktrap, louvain and leiden. See the OSCA book for details of others available in scran.
3.4 Modularity
Several methods to detect clusters (‘communities’) in networks rely on a metric called “modularity”. For a given partition of cells into clusters, modularity measures how separated clusters are from each other, based on the difference between the observed and expected weight of edges between nodes. For the whole graph, the closer to 1 the better.
3.5 Pros and Cons of graph based clustering
- Pros:
- fast and memory efficient (avoids the need to construct a distance matrix for all pairs of cells)
- no assumptions on the shape of the clusters or the distribution of cells within each cluster
- no need to specify a number of clusters to identify (but the size of the neighbourhood used affects the size of clusters)
- Cons:
- loss of information beyond neighboring cells, which can affect community detection in regions with many cells.
4 Implementation
The implementation of clustering in R is carried out using functions
from a number of different packages, in particular the bluster
and igraph packages. scran provides a handy “wrapper”
function clusterCells that allows us use a variety of
different algorithms with one simple command.
By default clusterCells just returns a vector containing
the cluster number for each cell. We can also retrieve the intermediate
statistics (varying according to the algorithm used) and the SNN graph
by specifying the bluster argument full = TRUE. If
you are only interested in retrieving the clusters, this isn’t necessary
but in this first instance we will retrieve the graph and visualise it.
The default algorithm for clusterCells is Walktrap with k is set to 10
by default. The default edge weighting is “rank”.
clustering1 <- clusterCells(sce, use.dimred="corrected", full=TRUE)This has defined 24 clusters with varying numbers of cells:
table(clustering1$clusters)##
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
## 133 154 243 66 194 74 14 151 313 64 34 143 204 498 844 31 80 24 56 56
## 21 22 23 24
## 67 17 24 16The number of cells in the data set is large and plotting all the cells would take too long, so we randomly choose 1000 nodes (cells) in the network before plotting the resulting smaller network. Adding sample data to the graph and plotting the results are done using the igraph package. Cells can be color-coded by sample type:
# extract the graph
snn.gr <- clustering1$objects$graph
# Add Sample group to vertices (nodes, ie cells)
V(snn.gr)$SampleGroup <- as.character(colData(sce)$SampleGroup)
# pick 1000 nodes randomly
set.seed(1423)
selectedNodes <- sample(3500, 1000)
# subset graph for these 1000 randomly chosen nodes
snn.gr.subset <- subgraph(snn.gr, selectedNodes)
# set colors for clusters
grps <- V(snn.gr.subset)$SampleGroup
cols <- c("dodgerblue", "lightyellow")[as.numeric(factor(grps))]
names(cols) <- grps
# plot graph
plot.igraph(snn.gr.subset,
layout = layout_with_fr(snn.gr.subset),
vertex.size = 3,
vertex.label = NA,
vertex.color = cols,
frame.color = cols,
main = "default parameters"
)
# add legend
legend('bottomright',
legend=unique(names(cols)),
pch=21,
pt.bg=unique(cols),
pt.cex=1, cex=.6, bty="n", ncol=1)
More commonly we will visualise the clusters by superimposing them on
a t-SNE or UMAP plot. We can store the clusters in the sce
object colData.
sce$Clusters1 <- clustering1$clusters
plotReducedDim(sce,
dimred = "TSNE_corrected",
colour_by="Clusters1",
text_by = "Clusters1")
5 The Walktrap method
The walktrap method relies on short random walks (a few steps) through the network. These walks tend to be ‘trapped’ in highly-connected regions of the network. Node similarity is measured based on these walks. Nodes are first each assigned their own community. Pairwise distances are computed and the two closest communities are grouped. These steps are repeated a given number of times to produce a dendrogram. Hierarchical clustering is then applied to the distance matrix. The best partition is that with the highest modularity. The original article describing the algorithm is Pons P, Latapy M (2006) Computing communities in large networks using random walks. J Graph Algorithms Appl 10(2):191–218
Walktrap is the default algorithm for clusterCells,
k is set to 10 by default and the default edge weighting is
“rank”. To explicitly request a specific algorithm and to set the
k to a different number of nearest neighbours, we use a
SNNGraphParam object from the bluster package
(which is the package clusterCells is using under the
hood).
Let’s set the k to 15 but keep the other parameters the same. This time we will just return the clusters:
sce$walktrap15 <- clusterCells(sce,
use.dimred = "corrected",
BLUSPARAM = SNNGraphParam(k = 15,
cluster.fun = "walktrap"))This time we have defined 16 clustering. As a general rule, increasing k will tend to decrease the number of clusters (not always, but generally).
table(sce$walktrap15)##
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
## 438 260 124 84 322 51 154 125 38 960 142 501 26 57 202 16We can visualise the assignment of cells from different samples to the clusters using a heatmap. This gives us an overview of how well each cluster is represented across the samples and the replicates. Several clusters (2, 8, 9 and 16) are present in the PBMMC samples, but absent from the ETV6_RUNX1 samples for instance.
w15_table <- log(table(sce$walktrap15, sce$SampleName)+1)
pheatmap(w15_table, cluster_rows = TRUE, cluster_cols = FALSE)
Most clusters comprise cells from several replicates of the same sample type, cluster 10 appears to be predominantly cells from the ETV6-RUNX samples.
We can visualise this on the TSNE:
plotReducedDim(sce,
dimred = "TSNE_corrected",
colour_by="walktrap15",
text_by = "walktrap15")
plotReducedDim(sce,
dimred = "TSNE_corrected",
colour_by="walktrap15",
text_by = "walktrap15",
other_fields = list("SampleGroup")) +
facet_wrap(vars(SampleGroup))
The different clustering algorithms may have additional parameters,
specific to the algorithm, that can be adjusted. With the walktrap
algorithm we could also tweak the number of “steps” in each walk. The
default is 4, but we could, for example, change this to 10 by adding the
parameter cluster.args = list(steps = 10) to the
SNNGraphParam object in the clusterCells
command.
6 The Louvain method
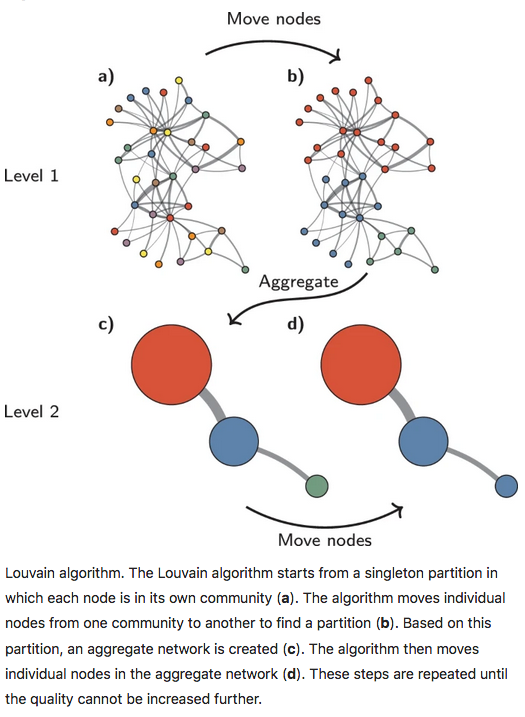
With the Louvain method, nodes are also first assigned their own community. This hierarchical agglomerative method then progresses in two-step iterations:
- nodes are re-assigned one at a time to the community for which they increase modularity the most, if at all.
- a new, ‘aggregate’ network is built where nodes are the communities formed in the previous step.
These two steps are repeated until modularity stops increasing. The diagram below is copied from this article.
We now apply the Louvain approach, store its outcome in the SCE object and show cluster sizes.
sce$louvain15 <- clusterCells(sce,
use.dimred = "corrected",
BLUSPARAM = SNNGraphParam(k = 15,
cluster.fun = "louvain"))table(sce$louvain15)##
## 1 2 3 4 5 6 7 8 9 10 11
## 396 57 884 445 213 157 496 265 111 214 262The t-SNE plot shows cells color-coded by cluster membership:
plotReducedDim(sce,
dimred = "TSNE_corrected",
colour_by = "louvain15",
text_by = "louvain15")
If we split by sample type we can see differences in the clusters between the sample groups:
plotReducedDim(sce,
dimred = "TSNE_corrected",
colour_by="louvain15",
text_by = "louvain15",
other_fields = list("SampleGroup")) +
facet_wrap(vars(SampleGroup))
7 The Leiden method
The Leiden method improves on the Louvain method by guaranteeing that at each iteration clusters are connected and well-separated. The method includes an extra step in the iterations: after nodes are moved (step 1), the resulting partition is refined (step2) and only then the new aggregate network made, and refined (step 3). The diagram below is copied from this article.

First run the clustering with clusterCells:
sce$leiden20 <- clusterCells(sce,
use.dimred = "corrected",
BLUSPARAM = SNNGraphParam(k = 20,
cluster.fun = "leiden"))We can quickly look at the results by summarising using
table.
table(sce$leiden20)##
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
## 341 61 932 428 210 508 3 1 147 120 116 140 214 262 17There are 15 clusters, although cluster 7 contains only 3 cells and cluster 8 contains only 1 cell.
The t-SNE plot shows cells color-coded by cluster membership:
plotReducedDim(sce,
dimred = "TSNE_corrected",
colour_by = "leiden20",
text_by = "leiden20")
8 Assessing cluster behaviour
A variety of metrics are available to aid us in assessing the behaviour of a particular clustering method on our data. These can help us in assessing how well defined different clusters within a single clustering are in terms of the relatedness of cells within the cluster and the how well separated that cluster is from cells in other clusters, and to compare the results of different clustering methods or parameter values (e.g. different values for k).
We will consider “Silhouette width” and “Modularity”. Further details and other metrics are described in the “Advanced” section of the OSCA book.
8.1 Silhouette width
The silhouette width (so named after the look of the traditional graph for plotting the results) is a measure of how closely related cells within cluster are to one another versus how closely related cells in the cluster are to cells in other clusters. This allows us to assess cluster separation.
For each cell in the cluster we calculate the the average distance to all other cells in the cluster and the average distance to all cells not in the cluster. The cells silhouette width is the difference between these divided by the maximum of the two values. Cells with a large silhouette are strongly related to cells in the cluster, cells with a negative silhouette width are more closely related to other clusters.
We will use the approxSilhouette function from the
bluster package. The resulting table gives us the silhouette
width for each cell, the cluster it belongs to, and which other cluster
it is most closely related to.
sil.approx <- approxSilhouette(reducedDim(sce, "corrected"),
clusters=sce$leiden20)
sil.approx## DataFrame with 3500 rows and 3 columns
## cluster other width
## <factor> <factor> <numeric>
## 1_CGACTTCGTCCAGTTA-1 1 3 0.0269813
## 1_AGAATAGCATACGCTA-1 1 3 0.0262305
## 1_TGACTAGAGAACTCGG-1 1 3 0.2867146
## 1_CTTAACTGTTATGCGT-1 2 8 0.3405912
## 1_CCCAGTTTCAAGCCTA-1 3 1 0.2456149
## ... ... ... ...
## 11_CCCAGTTCACATCCGG-1 14 7 0.0653069
## 11_TTAGTTCGTTAAAGAC-1 13 9 0.5494922
## 11_GATGAAATCTGGGCCA-1 6 9 0.4776268
## 11_ACGGAGAGTTAAGATG-1 13 1 0.2665459
## 11_TGGCGCAAGCGTTGCC-1 13 1 0.5182592We can view the results in as a beeswarm plot. We colour each cell according to either its current cluster or, if the cell has a negative silhouette width, the cluster that it is closest to.
plotSilBeeswarm <- function(silDat){
silTab <- silDat %>%
as.data.frame() %>%
mutate(closestCluster = ifelse(width > 0, cluster, other) %>% factor())
plt <- silTab %>%
ggplot(aes(x=cluster, y=width, colour=closestCluster)) +
ggbeeswarm::geom_quasirandom(method="smiley", alpha=0.6) +
theme_bw()
plt <- scater:::.resolve_plot_colours(plt,
silTab$closestCluster,
"closestCluster",
colour=TRUE)
plt
}
p1 <- plotSilBeeswarm(sil.approx)
p2 <- plotReducedDim(sce,
dimred = "TSNE_corrected",
colour_by="leiden20",
text_by = "leiden20")
p1 + p2
We could also look at the correspondence between different clusters by plotting these numbers on a grid showing for each cluster number of cells in that cluster that are closer to another cluster, colouring each tile by the proportion of the total cells in the cluster that it contains. Ideally we would like to see a strong diagonal band and only a few off-diagonal tiles containing small number of cells.
plotSilGrid <- function(silDat){
silDat %>%
as.data.frame() %>%
mutate(closestCluster = ifelse(width > 0, cluster, other) %>% factor()) %>%
count(cluster, closestCluster, name="olap") %>%
group_by(cluster) %>%
mutate(total = sum(olap)) %>%
mutate(proportion = olap / total) %>%
mutate(proportion = ifelse(cluster == closestCluster, proportion, -proportion)) %>%
ggplot(aes(x = cluster, y = closestCluster)) +
geom_tile(aes(fill = proportion)) +
geom_text(aes(label = olap), size=5) +
scale_fill_gradientn(colors = c("#fc8d59", "#ffffbf", "#91cf60"),
limits = c(-1, 1)) +
geom_vline(xintercept=seq(0.5, 30.5, by=1)) +
geom_hline(yintercept=seq(0.5, 30.5, by=1), colour="lightgrey", linetype=2) +
guides(fill = "none") +
theme(
aspect.ratio = 1,
panel.background = element_blank())
}
plotSilGrid(sil.approx)
From these two plots we can see that clusters 7, 8 and 12 appear to have a good degree of separation, however, clusters 7 and 8 only contains few cells, whilst there are many cells in other clusters that appear closer to them than they are to their assigned cluster. Perhaps clusters 7 and 8 needs to be merged with cluster 1.
Let’s do the same plots with the walktrap clusters generated with k=15.
sil.approx <- approxSilhouette(reducedDim(sce, "corrected"),
clusters=sce$walktrap15)
wp1 <- plotSilBeeswarm(sil.approx)
wp2 <- plotReducedDim(sce,
dimred = "TSNE_corrected",
colour_by="walktrap15",
text_by = "walktrap15")
wp3 <- plotSilGrid(sil.approx)
wp1 + wp2 + wp3
This clustering appears to have generated a set of clusters with slightly better separatedness than the Leiden method with a k of 20.
And again with the louvain clusters:
sil.approx <- approxSilhouette(reducedDim(sce, "corrected"),
clusters=sce$louvain15)
lp1 <- plotSilBeeswarm(sil.approx)
lp2 <- plotReducedDim(sce,
dimred = "TSNE_corrected",
colour_by="louvain15",
text_by = "louvain15")
lp3 <- plotSilGrid(sil.approx)
lp1 + lp2 + lp3
There seems to be a greater degree of overlap between these clusters, perhaps more resolution would improve this clustering - we might consider reducing k.
8.2 Modularity to assess clusters quality
As mentioned earlier, the modularity metric is used in evaluating the
separatedness of clusters. Some of the clustering algorithms,
e.g. Louvain, seek to optimise this for the entire NN graph as part of
their cluster detection. Modularity is a ratio between the observed
weights of the edges within a cluster versus the expected weights if the
edges were randomly distributed between all nodes. Rather than
calculating a single modularity value for the whole graph, we can
instead calculate a pair-wise modularity value between each pair of
clusters using the pairwiseModularity function from the
bluster package. For this we need to have the graph from the
clustering, so we will rerun the walktrap clustering with k=15 to obtain
this. We can plot the resulting ratios on a heatmap. We would expect the
highest modularity values to be on the diagonal.
walktrap15 <- clusterCells(sce,
use.dimred = "corrected",
BLUSPARAM = SNNGraphParam(k = 15,
cluster.fun = "walktrap"),
full = TRUE)
g <- walktrap15$objects$graph
ratio <- pairwiseModularity(g, walktrap15$clusters, as.ratio=TRUE)
hm1 <- pheatmap(log2(ratio+1),
cluster_rows=FALSE,
cluster_cols=FALSE,
color=colorRampPalette(c("white", "blue"))(100))
We can compare this to the silhouette width grid
wp4 <- ggplotify::as.ggplot(hm1)
wp2 + wp3 + wp4
Largely, this reflects what we saw from the silhouette widths, but also reveals some additional inter-connectedness between other clusters.
8.3 Comparing two sets of clusters
We can assess the concordance between different clustering methods to get a better idea of how they eachtreat the data, e.g. does one cluster from one method equate to just one cluster in the other or is it a combination of different clusters. This may be revealing about the underlying biology. We will use the Jaccard index as measure of concordance between clusters. A value of 1 represents perfect concordance between clusters (i.e. they contain exactly the same cells).
jacc.mat <- linkClustersMatrix(sce$louvain15, sce$walktrap15)
rownames(jacc.mat) <- paste("Louvain", rownames(jacc.mat))
colnames(jacc.mat) <- paste("Walktrap", colnames(jacc.mat))
pheatmap(jacc.mat, color=viridis::viridis(100), cluster_cols=FALSE, cluster_rows=FALSE)
We can see that Louvain clusters 2, 6, 7, and 9 are equivalent to walktrap clusters 14, 7, 12, and 11 respectively. The remaining Louvain clusters are combinations of cells from various walktrap clusters. We may want to look at marker genes for these clusters to assess what these two different views are telling us about the biology.
9 Cluster sweep
As we have seen, there are a number of different parameters we can
change to alter the final clustering result - primarily the k
used to build the NN graph, the edge weighting method and the clustering
algorithm. There is no one gold standard that will fit all data, so, in
most cases, it is necessary to assess a number of different clusterings
to obtain one that provides a view of the data that suits our biological
interpretations. The clusterSweep function allows us to
apply a range of different parameters in one go and obtain the
clustering for each.
For example, suppose we wish to assess the effect of different values of k on the walktrap clustering. We can parallelize this process to make it faster.
out <- clusterSweep(reducedDim(sce, "corrected"),
BLUSPARAM = NNGraphParam(),
k = as.integer(c(5, 10, 15, 20, 25)),
cluster.fun = "walktrap",
BPPARAM=BiocParallel::MulticoreParam(7))The resulting object is a list containing a DataFrame with the clusters for each combination of the clustering parameters and a corresponding DataFrame showing the parameters used to generate each of these:
out$clusters[,1:4]## DataFrame with 3500 rows and 4 columns
## k.5_cluster.fun.walktrap k.10_cluster.fun.walktrap
## <factor> <factor>
## 1 19 9
## 2 19 9
## 3 19 9
## 4 27 20
## 5 8 15
## ... ... ...
## 3496 9 5
## 3497 15 1
## 3498 20 14
## 3499 3 16
## 3500 15 1
## k.15_cluster.fun.walktrap k.20_cluster.fun.walktrap
## <factor> <factor>
## 1 10 7
## 2 5 7
## 3 5 7
## 4 14 10
## 5 10 9
## ... ... ...
## 3496 2 3
## 3497 8 1
## 3498 12 8
## 3499 9 5
## 3500 8 1out$parameters## DataFrame with 5 rows and 2 columns
## k cluster.fun
## <integer> <character>
## k.5_cluster.fun.walktrap 5 walktrap
## k.10_cluster.fun.walktrap 10 walktrap
## k.15_cluster.fun.walktrap 15 walktrap
## k.20_cluster.fun.walktrap 20 walktrap
## k.25_cluster.fun.walktrap 25 walktrapWe can then combine this cluster sweep with the metrics for assessing cluster behaviour in order to get a overview of the effects of these parameter changes that may enable us to make some decisions as to which clustering or clusterings we may wish to investigate further.
Here we will just look at the mean silhouette width and the number of clusters.
df <- as.data.frame(out$parameters)
# get the number of clusters
df$num.clusters <- apply(out$clusters, 2, max)
# get the mean silhouette width
getMeanSil <- function(cluster) {
sil <- approxSilhouette(reducedDim(sce, "corrected"), cluster)
mean(sil$width)
}
df$silhouette <- map_dbl(as.list(out$clusters), getMeanSil)
nclPlot <- ggplot(df, aes(x = k, y = num.clusters)) +
geom_line(lwd=2)
silPlot <- ggplot(df, aes(x = k, y = silhouette)) +
geom_line(lwd=2)
nclPlot + silPlot
Based on our previous analysis and knowledge of the biology we may feel that 12 clusters represents a good number clusters, and we can see here that k = 25, provides this. Also k = 25 gives us a better silhouette score than lower values of k. On the other hand, perhaps k = 15 provides greater resolution of cell types (more clusters) with only a slight decrease in the silhouette score.
Earlier we looked at the Jaccard index as a means of comparing two different clusterings. We could apply the same method here:
jacc.mat <- linkClustersMatrix(out$clusters$k.15_cluster.fun.walktrap,
out$clusters$k.25_cluster.fun.walktrap)
rownames(jacc.mat) <- paste("Walktrap_15", rownames(jacc.mat))
colnames(jacc.mat) <- paste("Walktrap_25", colnames(jacc.mat))
pheatmap(jacc.mat,
color = viridis::viridis(100),
cluster_cols = FALSE,
cluster_rows = FALSE)
The OSCA book provides some additional methods for comparing different clusterings that can be combined with the cluster sweep results to assess cluster behaviour under different parameters.
In this section, we have just done a sweep changing the k, but it is also possible to combine this with multiple clustering algorithms and multiple edge weightings.
Note: In practice, on a full data set, with multiple algorithms and values of k, this can take a long time to run. Usually I do not run this interactively in R studio, but instead write an R script that will end by exporting the final output of clusterSweep to an RDS object which I can later load into R. This R script can then be sent as a job to the cluster. This is also means the job can be massively more parallelized by using more cores. We will see this in the exercise for this session.
10 Finalise clustering selection
When you have come to a decision about which clustering to use it is
convenient to add it to colData column called “label” using
the colLabels function. This means downstream code does not
need to be changed should you later decide to switch to a different
clustering, you’d simply need to change the contents of the “label”
column. This also makes the code easily re-usable for different
analyses.
For now we will use the walktrap k=15 clustering.
colLabels(sce) <- sce$walktrap1511 Expression of known marker genes
If we expect our clusters to represent known cell types for which there are well established marker genes, we can now start to investigate the clusters by plotting in parallel the expression of these genes. This can also help us in assessing if our clustering has satisfactorily partitioned our cells.
plotReducedDim(sce,
dimred = "TSNE_corrected",
colour_by = "label",
text_by = "label") +
ggtitle("Walktrap k=15 clusters")
Having identified clusters, we now display the level of expression of cell type marker genes to quickly match clusters with cell types. For each marker we will plot its expression on a t-SNE, and show distribution across each cluster on a violin plot.
We will be using gene symbols to identify the marker genes, so we
will switch the rownames in the SCE object to be gene symbols. We use
the scater function uniquifyFeatureNames to do this as
there are a few duplicated gene symbols.
rownames(sce) <- uniquifyFeatureNames(rowData(sce)$ID, rowData(sce)$Symbol)11.1 B-cells markers
Let’s look at the expression of the B cell marker CD79A.
plotReducedDim(sce,
dimred = "TSNE_corrected",
by_exprs_values = "logcounts",
colour_by = "CD79A",
text_by = "label")
plotExpression(sce,
exprs_values = "logcounts",
x = "label",
colour_by = "label",
features=c("CD79A"))
A large number of the clusters seem to represent B cells. This is what we expect from a cancer of B cells
11.2 Monocyte markers
We have a very clear cluster of monocytes
plotReducedDim(sce,
dimred = "TSNE_corrected",
by_exprs_values = "logcounts",
colour_by = "LYZ",
text_by = "label")
plotExpression(sce,
exprs_values = "logcounts",
x = "label",
colour_by = "label",
features=c("LYZ"))
11.3 Save data
Write SCE object to file.
saveRDS(sce, file="results/Caron_clustering_material.rds")11.4 Session information
devtools::session_info()## ─ Session info ───────────────────────────────────────────────────────────────
## setting value
## version R version 4.3.3 (2024-02-29)
## os Ubuntu 22.04.4 LTS
## system x86_64, linux-gnu
## ui X11
## language (EN)
## collate en_US.UTF-8
## ctype en_US.UTF-8
## tz Etc/UTC
## date 2024-04-08
## pandoc 3.1.12.3 @ /opt/miniforge3/envs/R/bin/ (via rmarkdown)
##
## ─ Packages ───────────────────────────────────────────────────────────────────
## package * version date (UTC) lib source
## abind 1.4-5 2016-07-21 [2] CRAN (R 4.3.0)
## beachmat 2.18.0 2023-10-24 [2] Bioconductor
## beeswarm 0.4.0 2021-06-01 [2] CRAN (R 4.3.0)
## Biobase * 2.62.0 2023-10-24 [2] Bioconductor
## BiocGenerics * 0.48.1 2023-11-01 [2] Bioconductor
## BiocNeighbors 1.20.0 2023-10-24 [2] Bioconductor
## BiocParallel 1.36.0 2023-10-24 [2] Bioconductor
## BiocSingular 1.18.0 2023-10-24 [2] Bioconductor
## bitops 1.0-7 2021-04-24 [2] CRAN (R 4.3.0)
## bluster * 1.12.0 2023-10-24 [2] Bioconductor
## bslib 0.6.1 2023-11-28 [2] CRAN (R 4.3.2)
## cachem 1.0.8 2023-05-01 [2] CRAN (R 4.3.0)
## cli 3.6.2 2023-12-11 [2] CRAN (R 4.3.2)
## cluster * 2.1.6 2023-12-01 [2] CRAN (R 4.3.2)
## codetools 0.2-19 2023-02-01 [2] CRAN (R 4.3.0)
## colorspace 2.1-0 2023-01-23 [2] CRAN (R 4.3.0)
## cowplot 1.1.3 2024-01-22 [2] CRAN (R 4.3.2)
## crayon 1.5.2 2022-09-29 [2] CRAN (R 4.3.0)
## DelayedArray 0.28.0 2023-10-24 [2] Bioconductor
## DelayedMatrixStats 1.24.0 2023-10-24 [2] Bioconductor
## devtools 2.4.5 2022-10-11 [2] CRAN (R 4.3.0)
## digest 0.6.35 2024-03-11 [2] CRAN (R 4.3.3)
## dplyr * 1.1.4 2023-11-17 [2] CRAN (R 4.3.2)
## dqrng 0.3.2 2023-11-29 [2] CRAN (R 4.3.2)
## DT * 0.32 2024-02-19 [2] CRAN (R 4.3.2)
## edgeR 4.0.2 2023-11-19 [2] Bioconductor 3.18 (R 4.3.2)
## ellipsis 0.3.2 2021-04-29 [2] CRAN (R 4.3.0)
## evaluate 0.23 2023-11-01 [2] CRAN (R 4.3.1)
## fansi 1.0.6 2023-12-08 [2] CRAN (R 4.3.2)
## farver 2.1.1 2022-07-06 [2] CRAN (R 4.3.0)
## fastmap 1.1.1 2023-02-24 [2] CRAN (R 4.3.0)
## forcats * 1.0.0 2023-01-29 [2] CRAN (R 4.3.0)
## fs 1.6.3 2023-07-20 [2] CRAN (R 4.3.1)
## generics 0.1.3 2022-07-05 [2] CRAN (R 4.3.0)
## GenomeInfoDb * 1.38.1 2023-11-08 [2] Bioconductor
## GenomeInfoDbData 1.2.11 2024-03-19 [2] Bioconductor
## GenomicRanges * 1.54.1 2023-10-29 [2] Bioconductor
## ggbeeswarm 0.7.2 2023-04-29 [2] CRAN (R 4.3.0)
## ggplot2 * 3.5.0 2024-02-23 [2] CRAN (R 4.3.2)
## ggrepel 0.9.5 2024-01-10 [2] CRAN (R 4.3.2)
## glue 1.7.0 2024-01-09 [2] CRAN (R 4.3.2)
## gridExtra 2.3 2017-09-09 [2] CRAN (R 4.3.0)
## gtable 0.3.4 2023-08-21 [2] CRAN (R 4.3.1)
## highr 0.10 2022-12-22 [2] CRAN (R 4.3.0)
## hms 1.1.3 2023-03-21 [2] CRAN (R 4.3.0)
## htmltools 0.5.7 2023-11-03 [2] CRAN (R 4.3.2)
## htmlwidgets 1.6.4 2023-12-06 [2] CRAN (R 4.3.2)
## httpuv 1.6.14 2024-01-26 [2] CRAN (R 4.3.2)
## igraph * 2.0.2 2024-02-17 [2] CRAN (R 4.3.2)
## IRanges * 2.36.0 2023-10-24 [2] Bioconductor
## irlba 2.3.5.1 2022-10-03 [2] CRAN (R 4.3.3)
## jquerylib 0.1.4 2021-04-26 [2] CRAN (R 4.3.0)
## jsonlite 1.8.8 2023-12-04 [2] CRAN (R 4.3.2)
## knitr * 1.45 2023-10-30 [2] CRAN (R 4.3.2)
## labeling 0.4.3 2023-08-29 [2] CRAN (R 4.3.1)
## later 1.3.2 2023-12-06 [2] CRAN (R 4.3.2)
## lattice 0.22-5 2023-10-24 [2] CRAN (R 4.3.1)
## lifecycle 1.0.4 2023-11-07 [2] CRAN (R 4.3.2)
## limma 3.58.1 2023-10-31 [2] Bioconductor
## locfit 1.5-9.9 2024-03-01 [2] CRAN (R 4.3.3)
## lubridate * 1.9.3 2023-09-27 [2] CRAN (R 4.3.1)
## magrittr 2.0.3 2022-03-30 [2] CRAN (R 4.3.0)
## Matrix 1.6-5 2024-01-11 [2] CRAN (R 4.3.2)
## MatrixGenerics * 1.14.0 2023-10-24 [2] Bioconductor
## matrixStats * 1.2.0 2023-12-11 [2] CRAN (R 4.3.2)
## memoise 2.0.1 2021-11-26 [2] CRAN (R 4.3.0)
## metapod 1.10.0 2023-10-24 [2] Bioconductor
## mime 0.12 2021-09-28 [2] CRAN (R 4.3.0)
## miniUI 0.1.1.1 2018-05-18 [2] CRAN (R 4.3.0)
## munsell 0.5.0 2018-06-12 [2] CRAN (R 4.3.0)
## patchwork * 1.2.0 2024-01-08 [2] CRAN (R 4.3.2)
## pheatmap * 1.0.12 2019-01-04 [2] CRAN (R 4.3.0)
## pillar 1.9.0 2023-03-22 [2] CRAN (R 4.3.0)
## pkgbuild 1.4.4 2024-03-17 [2] CRAN (R 4.3.3)
## pkgconfig 2.0.3 2019-09-22 [2] CRAN (R 4.3.0)
## pkgload 1.3.4 2024-01-16 [2] CRAN (R 4.3.2)
## profvis 0.3.8 2023-05-02 [2] CRAN (R 4.3.1)
## promises 1.2.1 2023-08-10 [2] CRAN (R 4.3.1)
## purrr * 1.0.2 2023-08-10 [2] CRAN (R 4.3.1)
## R6 2.5.1 2021-08-19 [2] CRAN (R 4.3.0)
## RColorBrewer 1.1-3 2022-04-03 [2] CRAN (R 4.3.0)
## Rcpp 1.0.12 2024-01-09 [2] CRAN (R 4.3.2)
## RCurl 1.98-1.14 2024-01-09 [2] CRAN (R 4.3.2)
## readr * 2.1.5 2024-01-10 [2] CRAN (R 4.3.2)
## remotes 2.5.0 2024-03-17 [2] CRAN (R 4.3.3)
## rlang 1.1.3 2024-01-10 [2] CRAN (R 4.3.2)
## rmarkdown 2.26 2024-03-05 [2] CRAN (R 4.3.3)
## rstudioapi 0.15.0 2023-07-07 [2] CRAN (R 4.3.0)
## rsvd 1.0.5 2021-04-16 [2] CRAN (R 4.3.0)
## S4Arrays 1.2.0 2023-10-24 [2] Bioconductor
## S4Vectors * 0.40.2 2023-11-23 [2] Bioconductor 3.18 (R 4.3.2)
## sass 0.4.9 2024-03-15 [2] CRAN (R 4.3.3)
## ScaledMatrix 1.10.0 2023-10-24 [2] Bioconductor
## scales 1.3.0 2023-11-28 [2] CRAN (R 4.3.2)
## scater * 1.30.1 2023-11-16 [2] Bioconductor
## scran * 1.30.0 2023-10-24 [2] Bioconductor
## scuttle * 1.12.0 2023-10-24 [2] Bioconductor
## sessioninfo 1.2.2 2021-12-06 [2] CRAN (R 4.3.0)
## shiny 1.8.0 2023-11-17 [2] CRAN (R 4.3.2)
## SingleCellExperiment * 1.24.0 2023-10-24 [2] Bioconductor
## SparseArray 1.2.2 2023-11-07 [2] Bioconductor
## sparseMatrixStats 1.14.0 2023-10-24 [2] Bioconductor
## statmod 1.5.0 2023-01-06 [2] CRAN (R 4.3.0)
## stringi 1.8.3 2023-12-11 [2] CRAN (R 4.3.2)
## stringr * 1.5.1 2023-11-14 [2] CRAN (R 4.3.2)
## SummarizedExperiment * 1.32.0 2023-10-24 [2] Bioconductor
## tibble * 3.2.1 2023-03-20 [2] CRAN (R 4.3.0)
## tidyr * 1.3.1 2024-01-24 [2] CRAN (R 4.3.2)
## tidyselect 1.2.0 2022-10-10 [2] CRAN (R 4.3.0)
## tidyverse * 2.0.0 2023-02-22 [2] CRAN (R 4.3.0)
## timechange 0.3.0 2024-01-18 [2] CRAN (R 4.3.2)
## tzdb 0.4.0 2023-05-12 [2] CRAN (R 4.3.0)
## urlchecker 1.0.1 2021-11-30 [2] CRAN (R 4.3.0)
## usethis 2.2.3 2024-02-19 [2] CRAN (R 4.3.2)
## utf8 1.2.4 2023-10-22 [2] CRAN (R 4.3.1)
## vctrs 0.6.5 2023-12-01 [2] CRAN (R 4.3.2)
## vipor 0.4.7 2023-12-18 [2] CRAN (R 4.3.2)
## viridis 0.6.5 2024-01-29 [2] CRAN (R 4.3.2)
## viridisLite 0.4.2 2023-05-02 [2] CRAN (R 4.3.0)
## withr 3.0.0 2024-01-16 [2] CRAN (R 4.3.2)
## xfun 0.42 2024-02-08 [2] CRAN (R 4.3.2)
## xtable 1.8-4 2019-04-21 [2] CRAN (R 4.3.0)
## XVector 0.42.0 2023-10-24 [2] Bioconductor
## yaml 2.3.8 2023-12-11 [2] CRAN (R 4.3.2)
## zlibbioc 1.48.0 2023-10-24 [2] Bioconductor
##
## [1] /Users/sawle01/R/x86_64-conda-linux-gnu-library/4.3
## [2] /opt/miniforge3/envs/R/lib/R/library
##
## ──────────────────────────────────────────────────────────────────────────────